After working for years within the small community of scientists studying rare genetic disorders, Prof. Anthony Futerman today finds himself invited to large scientific meetings. This is because the genetic defects behind Gaucher’s disease, the object of Futerman’s study, have recently been linked to Parkinson’s disease. “For a quarter of a century, I’ve been working on this rare inherited disorder that hardly anyone cares about, and now it turns out that my lab’s research may be relevant to the health of many more people than we once thought,” says Futerman, of the Weizmann Institute of Science’s Biomolecular Sciences Department.

(l-r) Dr. Natalia Santos Ferreira, Prof. Anthony Futerman and Dr. Shifra Ben-Dor suggest an explanation for the connection between two diseases
A few years ago some scientists had discovered a puzzling link: Mutations in the GBA1gene, which cause Gaucher’s disease, are also the most frequent genetic risk factor for Parkinson’s disease. On the face of it, Gaucher’s and Parkinson’s have nothing in common. In Gaucher’s, mutations in the GBA1 gene interfere with the breakdown of fatty molecules, causing them to accumulate in cells throughout the body and leading to an enlargement of the liver and spleen. Most patients have a mild form of Gaucher’s, but even in the more severe forms that affect the brain, the neurological symptoms are different from those of Parkinson’s, which causes tremors and changes in speech and gait.
The new study in Futerman’s lab, published in Cell Reports, might help explain the genetic connection between the two diseases. The study may also help predict the course of Gaucher’s and lead to a potential treatment for its most debilitating symptoms.
In the study, postdoctoral fellow Dr. Andrés D. Klein and other team members induced Gaucher’s disease in fifteen strains of mice. Survival rates varied enormously in the different strains: Some of the mice came down with a severe form of the disease and died soon, whereas others had a milder form and lived much longer. By performing a genetic analysis, the scientists identified nearly 20 genes that appeared to affect the severity of the disease. If these findings are confirmed in humans, analysis of the modifier genes may help predict the course of Gaucher’s and explain why it varies so widely in different people.
The study may also help explain why some people with the genetic features of Gaucher’s have no symptoms at all. Even though it takes two defective copies of the gene, one inherited from each parent, for the disease to develop, mutations in the GBA1 geneoccur relatively frequently in a few ethnic groups. In particular, about one in ten Ashkenazi Jews is a carrier of the faulty GBA1 gene, so one would expect Gaucher’s to be much more prevalent among this group than it actually is. “It’s possible that some people carry two defective copies of the GBA1, but the modifier genes we’ve discovered prevent the disease from manifesting itself,” says Futerman.
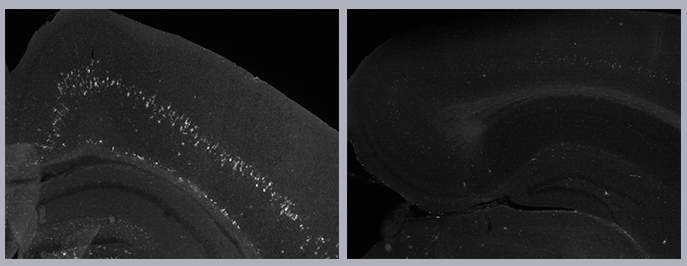
Among the mice with Gaucher’s disease, those with signs of inflammation in the brain (white marks, left) had significantly shorter lifespans than mice without such inflammation (right)
The scientists further found that they could delay Gaucher’s disease in mice by targeting one of the modifier genes that is known to play a role in signal transmission in the brain with memantine, a drug for neurodegenerative diseases. If the same results are produced in patients with Gaucher’s, in may be possible to treat some of its neurological symptoms.
In fact, the potential explanation for the link to Parkinson’s, a neurodegenerative disorder, emerged when the scientists realized that a large proportion of the newly identified modifier genes are involved in neurological function. “These genes may help uncover biochemical processes that are common to both diseases, and this, in turn, might lead to the missing link between Gaucher’s and the risk of Parkinson’s disease,” says Futerman.
The modifier genes we’ve discovered prevent the disease from manifesting itself
There’s also indirect evidence pointing to shared biochemical processes. These may have to do with the lysosome, the organelle that degrades unwanted cellular material. In Gaucher’s, the fatty molecules that aren’t broken down properly accumulate in this organelle. The lysosome is also known to become less effective in older age — a time of life when Parkinson’s disease most commonly develops.
Further study of Gaucher’s modifier genes may also shed new light on other genetic disorders in which fatty molecules abnormally accumulate in the lysosome, including Tay-Sachs disease.
Futerman’s team included postdoctoral fellow Dr. Natalia Santos Ferreira and staff scientist Dr. Shifra Ben-Dor of Weizmann’s Life Sciences Core Facilities. The study was conducted in collaboration with Prof. Alfred Merrill and Dr. Jingjing Duan of the Georgia Institute of Technology in Atlanta, Georgia, Prof. John Hardy of University College London and Prof. Timothy Cox of the University of Cambridge.
Prof. Anthony H. Futerman’s research is supported by the estate of Lilly Fulop. Prof. Futerman is the incumbent of the Joseph Meyerhoff Professorial Chair of Biochemistry.
This news appaered on Weizmann.ac.il
Recent Comments